

Recent progress in quantum chemistry has been accelerated by means of quantum computing. Within the framework of Quantum-Centric Supercomputing, hybrid quantum-classical algorithms efficiently compute properties of matter at a scale beyond brute-force classical methods. Typically, quantum chemistry simulations are based on the second-quantized electronic Hamiltonian within the Born-Oppenheimer approximation. This means calculating the one- and two-body integrals using anvapproximate (classical) method, such as Hartree-Fock, given the atomic positions within the unit cell.
In contrast, lattice Hamiltonians are rarely used in the context of chemistry. However, they are promising for applications involving Artificial Intelligence (AI) methods, as they can effectively encode the entire electronic structure within a limited number of parameters.
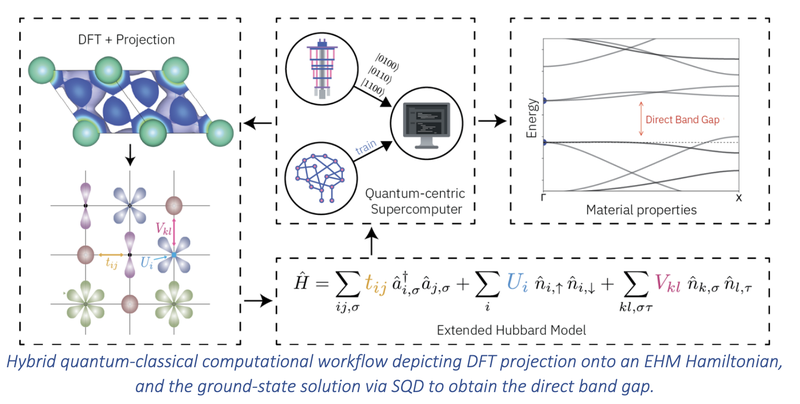
In this work, we investigate the band gap of periodic materials in a lattice Hamiltonian representation by means of a hybrid, quantum-classical computational workflow. Specifically, we employ the Extended Hubbard Model (EHM), which parameterizes the electronic Hamiltonian using a set of {tij} hopping parameters, as well as {Ui} intra-site and {Vij} inter-site Coulomb interactions, to account for localization and hybridization effects. For each candidate material, we calculate the Hamiltonian parameters self-consistently within the Density Functional Theory (DFT), based on an atomic-level description of their unit cell. The electronic Hamiltonian is then solved using the Sample-based Quantum Diagonalization (SQD) framework. By applying this approach to materials with metal-oxide bonds, we compute key electronic properties, such as band gaps1. In addition, we investigate the potential of EHM-derived parameters as descriptors for AI-driven structure-property prediction models, harnessing machine learning to bridge the gap between quantum models and materialproperty trends.
1 Duriez, Alan, et al. "Computing band gaps of periodic materials via sample-based quantum diagonalization." arXiv preprint arXiv:2503.10901 (2025).